If you work in the medical device industry, you have probably heard the term “ISO 13485” thrown around in meetings, audit preparations, and regulatory discussions. But what does it actually take to build a medical device quality management system that meets this international standard? More importantly, how do you move from reading the requirements to living them every day on the production floor, in the design office, and during supplier evaluations?
This guide walks you through everything: the current ISO 13485:2016 revision, the full scope of ISO 13485 standards for medical devices, a clause‑by‑clause breakdown of ISO 13485 requirements, practical ISO 13485 guidelines for medical devices, a clear comparison of ISO 13485 vs. 21 CFR 820, documentation toolkits, consultant roles, and the certification process. No fluff. Just actionable insight.
The objective is simple: help manufacturers build a quality management system that is functional, audit-ready, and sustainable over time.
What is ISO 13485:2016?
ISO 13485 is an internationally recognized quality management system (QMS) standard created specifically for organizations operating within the medical device industry. It establishes requirements for processes that influence device safety, effectiveness, consistency, and regulatory compliance.
Back in 2016, the International Organization for Standardization released a fundamental update to the medical device QMS standard. The previous version from 2003 served the industry well for over a decade, but medical technology evolved faster than anyone expected. Software‑driven devices, combination products, and global supply chains demanded a more risk‑based, lifecycle‑oriented approach.
ISO 13485:2016 is not just a refresh – it is a complete rethinking. Where the 2003 version leaned heavily on ISO 9001’s customer satisfaction model, the 2016 version focuses squarely on regulatory compliance and product safety. That shift matters because notified bodies and regulators now expect you to prove not only that your customers are happy, but that your processes consistently deliver safe, effective devices.
Key Changes from Previous Versions
The 2016 revision introduced substantial changes intended to reflect modern manufacturing realities, global supply chains, software-driven devices, and increasing regulatory expectations. If you are migrating from the 2003 standard, watch for these four major changes:
- Risk‑Based Approach Throughout – Clause 4.1.2 requires you to apply risk management to all QMS processes, not just product design. That means purchasing, training, infrastructure, and even document control need risk assessments.
- Greater Emphasis on Software Validation – The 2016 version explicitly requires validation of any software used in the QMS (e.g., CAPA tracking, complaint handling) and software used in production or service provision.
- Tighter Control of Outsourced Processes – You cannot outsource your way out of responsibility. Clause 4.1.5 demands that you document controls for every outsourced process that affects product conformity.
- Strengthened Complaint Handling and Regulatory Reporting – Clause 8.2.2 and 8.3 now align more closely with global regulatory requirements (MDR, MDSAP, FDA). You need defined processes for feedback, complaints, and advisory notices.
Transition Timelines and Current Applicability
The transition period from ISO 13485:2003 to 13485:2016 ended in early 2019. Today, all new certifications and surveillance audits are conducted against the 2016 version. If your certificate still says 2003, it is no longer valid. For most organizations, the real question is not “Should we upgrade?” but “How do we maintain compliance with the 2016 version while preparing for the next wave of regulations (EU MDR, IVDR, FDA QMSR)?”
Good News: ISO 13485:2016 remains the gold standard for the foreseeable future. No major revision is announced yet, but keep an eye on harmonization efforts with FDA’s QMSR (which directly references ISO 13485).

ISO 13485 Standards for Medical Devices
Many people mistakenly think ISO 13485 stands alone. It does not. It is the umbrella standard for medical device QMS, but it relies on a family of supporting standards. Think of it like a toolbox:
Most manufacturers also rely on related standards and regulatory frameworks.
| Standard | Role |
|---|---|
| ISO 13485 | Quality Management System requirements |
| ISO 14971 | Risk management (mandatory reference) |
| ISO 13408 | Aseptic processing |
| ISO 14155 | Clinical investigations |
| ISO 15223 | Symbols for labeling |
| IEC 62304 | Medical device software lifecycle |
| IEC 60601 | Electrical safety |
When you claim conformity to ISO 13485, you implicitly agree to apply ISO 14971 for risk management. Not as a suggestion – as a requirement (see Clause 7.1). Similarly, your design and development process must consider applicable product‑specific standards.
Relationship with ISO 14971 and ISO 13458
Let us clear up a frequent confusion: ISO 13458 does not exist. It is a typo. People often write “ISO 13458” when they mean ISO 13485. If you see that in a document or email, politely correct it – it could cause confusion during an audit. Some may confuse it with ISO 13485:2016’s clause numbering, but no, there is no separate standard with that number.
What about ISO 14971? That is the real companion. ISO 14971:2019 (or the older 2007 version) gives you the formal process for hazard identification, risk estimation, risk control, and residual risk evaluation. ISO 13485 requires you to maintain a risk management file throughout the product lifecycle. In practice, most medical device companies integrate the two by referencing risk analysis outputs in design reviews, production process validations, and CAPA decisions.
Recognition by Regulatory Bodies (MDSAP, CE Mark, FDA)
Here is why ISO 13485 is not just a nice‑to‑have: regulators trust it.
- MDSAP (Medical Device Single Audit Program) – Canada, United States, Brazil, Australia, and Japan recognize ISO 13485 as the foundation. Passing an MDSAP audit can replace multiple country‑specific inspections.
- CE Marking – Under the EU MDR and IVDR, manufacturers must implement a QMS that conforms to ISO 13485 (though the regulations add extra requirements). Notified bodies almost always audit against ISO 13485 as part of CE certification.
- FDA – Historically the FDA followed 21 CFR 820 (Quality System Regulation). But in 2024, the FDA proposed the QMSR rule to align 21 CFR 820 with ISO 13485:2016. Once finalized, compliance with ISO 13485 will largely satisfy FDA’s device QMS requirements – a huge win for global manufacturers.
In Short: invest in ISO 13485, and you open doors to most major markets.

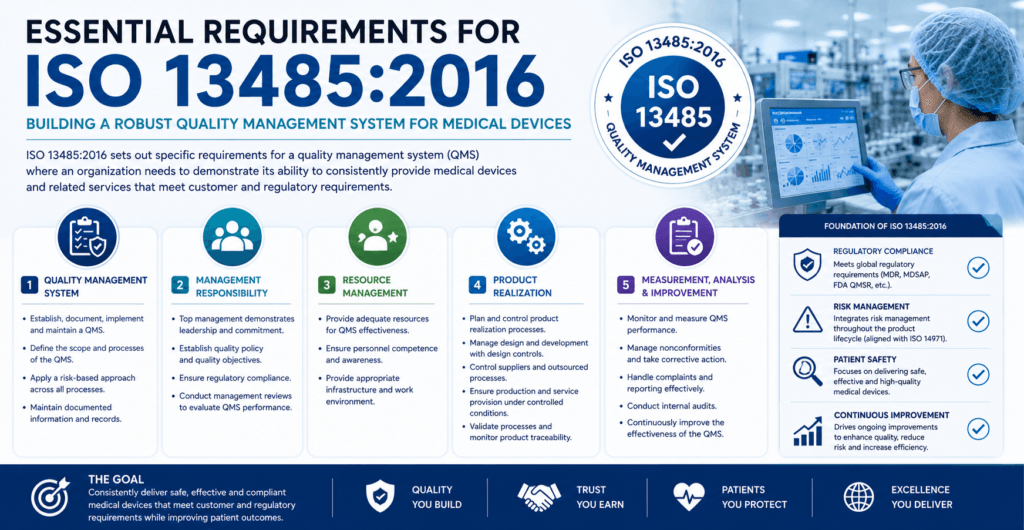
ISO 13485 Requirements – Complete Clause-by-Clause Breakdown
Let us walk through each of the eight main clauses. I will highlight what auditors actually look for and where most companies stumble.
Clause 4: Quality Management System – Documentation Toolkit Essentials
What it says: You must establish, document, implement, and maintain a QMS. Then you must continuously improve its effectiveness.
Key Sub‑Clauses:
- 4.1.2 – Apply a risk‑based approach to all QMS processes.
- 4.1.4 – Maintain a quality manual (scope, procedures, interactions of processes).
- 4.2 – Control of documents and records. Every document needs identification, approval, version control, and removal of obsolete copies.
Most Common Non‑Conformity: Missing records. Clause 4.2.5 lists required records (training, validation, design reviews, etc.). If you say you did something but have no evidence, it did not happen.
Clause 5: Management Responsibility
What it says: Top management must show commitment – not just on paper.
Expectations:
- 5.1 – Communicate the importance of regulatory and QMS requirements.
- 5.4.1 – Establish quality objectives (measurable!).
- 5.5.1 – Define responsibilities and authorities.
- 5.6 – Management review at planned intervals. Review inputs (audit results, feedback, process performance, changes) and outputs (decisions on improvements, resource needs).
Real‑World Tip: Do not let management review become a box‑ticking exercise. The best companies use it to challenge the QMS – “Why did we have three CAPAs in sterilization? Do we need retraining or a different supplier?”
Clause 6: Resource Management
What it says: Provide the people, infrastructure, and work environment needed for product quality.
- 6.2 – Human resources: define competence, provide training, evaluate effectiveness. Keep training records.
- 6.3 – Infrastructure: buildings, equipment, utilities, transport.
- 6.4 – Work environment: cleanroom controls, temperature, humidity, contamination prevention.
Hidden Trap: Competence evaluation is not just “he attended a class”. You need to show that the person can actually do the job – observation, testing, or practical demonstration.
Clause 7: Product Realization
This is the longest and most detailed clause. Break it down:
- 7.1 – Planning of product realization. Link to risk management.
- 7.2 – Customer‑related processes (determine requirements, review contracts, handle changes).
- 7.3 – Design and development. A full sub‑clause: planning, inputs, outputs, review, verification, validation, transfer, and control of changes. Design validation must be performed on the finished device under actual or simulated use conditions.
- 7.4 – Purchasing. Evaluate and re‑evaluate suppliers based on risk. Keep records.
- 7.5 – Production and service provision. Control conditions (documented procedures, monitoring, equipment validation). Also includes sterilization process validation and traceability (including Unique Device Identification – UDI).
- 7.6 – Control of monitoring and measuring equipment. Calibrate or verify at defined intervals.
What Auditors Drill Into: Design transfer (7.3.8) – did you successfully translate design outputs into production specifications? And supplier control (7.4) – do you have objective evidence of supplier approval?
Clause 8: Measurement, Analysis, and Improvement
The feedback loop that closes the QMS circle.
- 8.2 – Monitoring and measurement. Includes feedback from customers (complaints), internal audits, and process/product monitoring.
- 8.3 – Control of nonconforming product. Decide to rework, scrap, or accept by concession. Re‑validation required after rework.
- 8.4 – Analysis of data. Use statistical techniques where appropriate.
- 8.5 – Improvement. Corrective action (CAPA) for root cause elimination. Preventive action before problems occur.
Gold Nugget: Many companies confuse correction (fix the immediate issue) with corrective action (prevent recurrence). Auditors love to find CAPAs that stop at the first level. Always ask “Why?” five times.

Mandatory Documents and Records
ISO 13485:2016 requires a specific set of documented procedures and records. Here are the 21+ essential documents you must have:
- Quality manual (4.2.2)
- Document control procedure (4.2.4)
- Record control procedure (4.2.5)
- Training records (6.2)
- Infrastructure maintenance records (6.3)
- Work environment monitoring records (6.4)
- Design and development plan (7.3.1)
- Design input/output records (7.3.3/7.3.4)
- Design review records (7.3.5)
- Design verification/validation records (7.3.6/7.3.7)
- Design transfer records (7.3.8)
- Purchasing records (7.4)
- Production process validation records (7.5.6)
- Traceability records (7.5.9)
- Calibration records (7.6)
- Customer feedback/complaint records (8.2.1)
- Internal audit records (8.2.2)
- Monitoring and measurement records (8.2.3)
- Nonconforming product records (8.3)
- CAPA records (8.5.2/8.5.3)
Save Months of Work – Get the Complete Toolkit
Writing all these from scratch takes hundreds of hours. Instead, get the ISO 13485:2016 Documentation Toolkit with 122 ready‑to‑use documents – SOPs, templates, forms, validation protocols, risk management files, and more. Fully aligned with ISO 13485, ISO 14971, EU MDR, and FDA requirements. Editable, structured, and audit‑ready.
👉 Download the Toolkit Now and implement your QMS in weeks, not months.
Certification Process for ISO 13485:2016
Choosing a Certification Body
Only certain organizations can issue ISO 13485 certificates. For CE marking, you need a Notified Body under EU MDR (e.g., BSI, TÜV SÜD, DEKRA, SGS). For standalone ISO 13485 certification without CE marking, you can use any accredited certification body.
Factors to consider when choosing:
- Industry expertise – Do they have auditors who know your device type (active implantable, IVD, software, non‑sterile)?
- Global recognition – Is the certificate accepted in your target markets (e.g., Japan requires a specific accreditation)?
- Cost and schedule – Request quotes from three bodies. Stage 1 and Stage 2 audits typically cost between 5,000and15,000 depending on company size.
- Language and location – Choose an auditor who speaks your team’s language fluently. Miscommunication leads to unnecessary non‑conformities.
Stage 1 (Documentation) and Stage 2 (Implementation) Audits
Stage 1 – Readiness review (usually 1‑2 days on site or remote)
The auditor reviews your quality manual, key procedures, and documentation toolkit. They check for completeness against Clause 4.2. They also assess site‑specific issues (e.g., does your cleanroom match the drawings?). At the end, you get a report of potential non‑conformities. You fix those before Stage 2.
Typical Outcome: A few minor non‑conformities or observations. No major non‑conformities at Stage 1 is expected.
Stage 2 – Implementation verification (2‑5 days on site depending on size)
The auditor follows your product from design to delivery. They interview operators, review records (training, calibration, CAPA), and observe production. They check that what you wrote in your procedures actually happens on the floor.
Possible Outcomes:
- Recommend certification – No major non‑conformities. You close any minors within a defined time (usually 30‑60 days).
- Not recommend certification – One or more major non‑conformities (systemic breakdown, missing critical process). You must correct them and undergo a partial re‑audit.
Pro Tip: Do not schedule Stage 2 immediately after Stage 1. Leave at least 4‑6 weeks to implement any corrective actions. Rushing kills certification.
Surveillance and Recertification
Certification is not permanent. You earn it every year.
- Year 1 and 2 – Surveillance audits (usually 1‑2 days each). The auditor checks that your QMS continues to function and that you closed previous non‑conformities.
- Year 3 – Recertification audit (similar in scope to Stage 2). You need to demonstrate improvement over the three‑year period, not just maintenance.
Common Mistake: Letting the QMS decay between audits. Do not. Run your internal audits on schedule. Keep your management review alive. Then surveillance becomes a simple confirmation rather than a crisis.

Role of ISO 13485 Consultants
Some companies go it alone. Others bring in help. You might need a consultant if:
- You are a startup – No prior QMS experience, and you need certification before a funding or distribution milestone.
- Your previous certification lapsed – You let the QMS go stale and need to rebuild quickly.
- You received major non‑conformities – A certification audit or regulatory inspection revealed deep gaps that your internal team cannot fix.
- You are integrating ISO 13485 with other standards – For example, combining 13485 with ISO 9001 or with MDSAP requirements.
Warning: Do not hire a consultant to “write the QMS for you.” That never works because your staff will not understand or own it. The best consultants coach your team to write their own procedures.
How consultants help with ISO 13485 requirements, internal audits, and certification?
A good consultant provides four specific services:
- Gap Analysis and Remediation Planning – They will interview your team, review your existing documents, and produce a prioritized action plan. This alone often pays for itself because it stops you from wasting time on low‑impact tasks.
- Template Toolkit – Not a full QMS, but templates for procedures, forms, and audit checklists. You then customize them. Look for consultants who provide editable, device‑industry‑specific templates (not generic ISO 9001 templates).
- Internal Audit Support – They can perform a pre‑certification audit or train your internal auditors. The mock audit is especially valuable – it uncovers issues before the notified body arrives.
- Audit Coaching – They will sit with you during the certification audit (as a shadow) and help you respond to findings. Some consultants even negotiate minor non‑conformities into observations.
Red Flag: A consultant who promises certification “in 90 days or less.” Realistic timelines are 6‑9 months. Anything faster usually means shortcuts that will fail during surveillance.
ISO 13485 CONSULTANCY
Get a practical overview of the procedures, records, and validation activities required for compliance.

1. What is ISO 13485:2016?
Answer: ISO 13485:2016 is the international standard that specifies requirements for a quality management system specific to medical devices. Unlike ISO 9001, which focuses on customer satisfaction, ISO 13485 emphasizes regulatory compliance and product safety throughout the device lifecycle – from design and development to production, storage, and distribution. It is the most widely recognized QMS standard for medical device manufacturers worldwide.
2. What are the main ISO 13485 requirements for medical devices?
Answer: The standard is organized into eight clauses. The main operational requirements are: Clause 4 (QMS documentation and risk‑based processes), Clause 7 (product realization – design, purchasing, production), and Clause 8 (measurement, analysis, and improvement – including CAPA and complaint handling). You must also meet requirements for management responsibility (Clause 5) and resource management (Clause 6). Mandatory outputs include a quality manual, 21+ documented procedures, and records for design, training, validation, and corrective actions.
3. How do ISO 13485 guidelines for medical devices differ from FDA requirements?
Answer: Historically, FDA followed 21 CFR 820 (Quality System Regulation), which differed from ISO 13485 in areas like design validation, CAPA documentation, and complaint handling. However, in 2024 the FDA proposed the QMSR rule to align 21 CFR 820 with ISO 13485:2016. Once finalized, compliance with ISO 13485 will largely satisfy FDA requirements. Until then, minor differences remain – for example, FDA explicitly requires a complaint file and device history records, while ISO 13485 covers these indirectly. The best practice is to build your QMS around ISO 13485 and then add a short list of US‑specific procedures.
4. What is the difference between ISO 13485 vs. 21 CFR 820?
Answer: ISO 13485 is an international voluntary standard used for CE marking, MDSAP, and many national regulations. 21 CFR 820 is a US federal regulation enforced by the FDA. The two were not identical but largely overlapped. The main differences used to be in design controls (more prescriptive in FDA), CAPA (separate subpart in FDA), and complaint handling (explicit MDR linkage in FDA). With the FDA’s new QMSR rule, 21 CFR 820 will directly reference ISO 13485:2016, effectively harmonizing them. Going forward, a single QMS can satisfy both with minor supplements.
5. Can I use the same QMS for both ISO 13485 and FDA compliance?
Answer: Yes, absolutely. In fact, that is the recommended approach. Design your medical device quality management system around ISO 13485:2016. Then map each FDA requirement (21 CFR 820, and soon the QMSR) to your ISO‑based procedures. You will likely find 90‑95% overlap. For the remaining 5‑10%, add specific work instructions or policy notes. Use a cross‑reference matrix in your quality manual to show how you meet both. This unified system saves time, reduces audit fatigue, and simplifies internal training.
The medical device industry is moving toward stricter regulations and higher expectations. But that is not a burden – it is an opportunity. A robust QMS built on ISO 13485 standards and guidelines does not just satisfy auditors; it helps you catch defects earlier, respond to complaints faster, and deliver safer products to patients who depend on them.
Start today. One clause at a time.
Need a ready‑to‑use ISO 13485 documentation toolkit or a personalized implementation plan? Contact our team of ISO medical device specialists for a free 30‑minute discovery call.